
Living with Thalassemia : Northern California Comprehensive Thalassemia Center
Living with Thalassemia
Since thalassemia is a chronic illness, the key to successful management of the disease is the integration of psychological wellness and counseling along with medical care.
Health care providers will encounter many complex personal and cultural issues when caring for thalassemia patients and their families. These are our experiences.
In this section you will find information on what to expect from your child's development, what to expect as a patient and ways in which you can ease transitions.
Life Stages: Infancy
Most families find out about their child's diagnosis shortly after birth. Even for families who may be familiar with thalassemia, this can be a difficult time. Adjusting to a new diagnosis can be challenging. You will be getting lots of new information and meeting lots of new health care providers. Expect your child to reach normal developmental milestones.
What you can do:
- Use you support network.Talk with family members and friends who have been helpful in the past. You may even want to bring in part of your support network when you see the doctor. If you don't know someone with your child's disease, think about asking your doctor's office to introduce you to another family.
- It's okay to ask questions and to ask them more than once. Try to pay attention to how you manage information; some families prefer information in writing, some families prefer to hear less information at once. If you know your preference, share it with your health care providers.
- Take care of yourself. Your ability to take care of your child is directly related to how you are doing. Take time to relax, participate in fun activities, and enjoy the new member of your family.
Life Stages: Toddlerhood
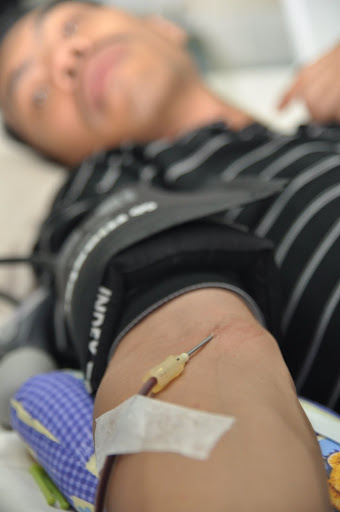
Children at this age are testing their environment for what they can do and testing their parents for what they are allowed to do. Children are not able to fully understand why they need to come to the hospital and why things that are uncomfortable are being done to them (needle sticks, exams). You may find increased resistance to invasive procedures such as blood draws or transfusions. If your child is on chronic transfusions, you may begin using Desferal at home during this time. It can be very difficult to stick your own child, both physically and emotionally.
If your child attends day care, you will need to think about what types of information you feel comfortable sharing about your child’s disease.
What you can do:
- Medical play can assist your child to master medical procedures. Have a child life specialist or psychosocial provider at your hospital work with your child around medical play. It can also be helpful to have a toy medical bag at home.
- During procedures or IV’s, help your child manage the stress by distracting them with books, songs or toys.
- Speak with your health care providers about what you should share with day care centers, baby sitters, etc.
- Continue to expect age appropriate behavior from your child and don’t be afraid to set limits.
Life Stages: School Years
Children during these years look for activities they are good at, math, reading, sports, art, helping around the house, etc. This need for a sense of mastery also extends to a child’s illness. Children will want to have more control over procedures and ask many more questions about why procedures and tests are being conducted. School age children being to look more towards their peers to assess their competence in academic and social arenas. Children will begin to take notice more that their peers do not come to the hospital like they do and will ask questions about this. As children get older, their cognitive development changes and they have a different understanding of themselves and their environment. You may get asked the same questions many times.
What you can do:
- Help your child find something that they are good at, encourage their interests.
- Try to give your child more choices, and therefore control, about their medical care. For example, give your child a choice about where the IV should be placed.
- Listen and take seriously your child’s questions about their illness and medical care. Answer questions as clearly and honestly as you can. When you are unsure of the answer or how to best talk with your child about a concern, talk to your health care provider.
Life Stages: Adolescence
Many parents and patients say that the teen years are the most difficult time for families. Families struggle with the shift of responsibility and control over the disease, academic progress, and social activities. As teens take more responsibility for their illness, compliance may become an issue. Teenagers spend less time at home, are more oriented toward their peers, and are motivated to be like their peers. All of these factors can lead towards non-compliant behavior. Adolescents begin to think more like adults and to understand adult concepts of illness and mortality. While teens can cognitively understand abstract concepts, many feel protected from negative consequences; this can lead towards risk-taking behaviors, including experimenting with drugs and alcohol, sex and aggressive behavior.
What you can do as a parent:
- Continue to negotiate with your child around disease responsibility.
- Have your teen start to develop independent relationships with his or her health care providers; this can help you negotiate the transfer of care to your child and give him/her a private and safe place to voice concerns.
- Get support and information from other parents going through this difficult time.
- Do not hesitate to ask for your family to meet with a psychosocial provider to help you manage these years.
What you can do as a patient:
- Try to find people to talk to who you think can listen to your questions or concerns about your disease. You may want to try friends, parents, relatives, health care providers, and other patients.
- Contact TAG (Thalassemia Action Group) at (800)935-0024 or (800)522-7222 (e-mail: ncaf@aol.com). This is a national peer support network of patients that provide information on thalassemia.
- Subscribe to a listserv (thalassemia@listbot.com). Join a virtual community of thalassemia patients or find a pen-pal.
Life Stages: Adulthood
The responsibilities of adult life can get even more complicated in the presence of a chronic illness. Adults face the challenge of maintaining relationships, work, medical insurance, and managing an often complicated medical disease. For some, reaching adulthood may also mean a change in their system of medical care from a pediatric setting to an adult setting. Compliance with medical management continues to be an issue as patients balance the issues of the use of invasive medical procedures and quality of life. Please read Proposal: Living as an Adult in Pediatric World in our Patient Forum Section
What you can do as an adult:
- Continue to get help and support from others, including your health care professionals, family and friends.
- If you have not already, contact the Thalassemia Action Group for support and to keep updated on new advances in thalassemia care. Phone: (800)935-0024 or (800)522-7222. Email: ncaf@aol.com.
- Work with a social worker or a legal action group to ensure that you know your rights in employment and insurance situations.
Contact Us
Northern California Comprehensive Thalassemia Center
Children's Hospital Oakland
Department of Hematology/Oncology
747 52nd Street, Oakland, CA 94609
Phone: 510-428-3885 x 5427
Email: info@thalassemia.com
Please note that due to liability issues, and in order to preserve the individual doctor-patient relationship, we cannot give medical advice on specific cases over the internet. Your physician can contact us directly if necessary.





































0 comments: