MedicineNet.com
•What is beta thalassemia?
•What is the difference between thalassemia minor and major?
•What is Mediterranean anemia?
•What is the genetic pattern of inheritance of beta thalassemia?
•The diagnosis of thalassemia major and minor
•The treatment of thalassemia major
The thalassemias are a group of genetic (inherited) blood disorders that share in common one feature, the defective production of hemoglobin, the protein that enables red blood cells to carry oxygen (and carbon dioxide). There are many different disorders with defective hemoglobin synthesis and, hence, many types of thalassemia.
The most familiar type of thalassemia is beta thalassemia. It involves decreased production of normal adult hemoglobin (Hb A), the predominant type of hemoglobin from soon after birth until death. (All hemoglobin consists of two parts: heme and globin). The globin part of Hb A has 4 protein sections called polypeptide chains. Two of these chains are identical and are designated the alpha chains. The other two chains are also identical to one another but differ from the alpha chains and are termed the beta chains. In persons with beta thalassemia, there is reduced or absent production of beta globin chains.
What is the difference between thalassemia minor and major?
There are two forms of beta thalassemia. They are thalassemia minor and thalassemia major (which is also called Cooley's anemia).
Thalassemia minor: The individual with thalassemia minor has only one copy of the beta thalassemia gene (together with one perfectly normal beta-chain gene). The person is said to be heterozygous for beta thalassemia.
Persons with thalassemia minor have (at most) mild anemia (with slight lowering of the hemoglobin level in the blood). This situation can very closely resemble that with mild iron-deficiency anemia. However, persons with thalassemia minor have a normal blood iron level (unless they have are iron deficient for other reasons). No treatment is necessary for thalassemia minor. In particular, iron is neither necessary nor advised.
Thalassemia major (Cooley's anemia): The child born with thalassemia major has two genes for beta thalassemia and no normal beta-chain gene. The child is homozygous for beta thalassemia. This causes a striking deficiency in beta chain production and in the production of Hb A. Thalassemia major is, therefore, a serious disease
.
The clinical picture associated with thalassemia major was first described in 1925 by the American pediatrician Thomas Cooley. Hence, the name Cooley's anemia in his honor.
At birth the baby with thalassemia major seems entirely normal. This is because the predominant hemoglobin at birth is still fetal hemoglobin (Hb F). Hb F has two alpha chains (like Hb A) and two gamma chains (unlike Hb A). It has no beta chains so the baby is protected at birth from the effects of thalassemia major.
Anemia begins to develop within the first months after birth. It becomes progressively more and more severe. The infant fails to thrive (to grow normally) and often has problems feeding (due to easy fatigue from lack of oxygen, with the profound anemia), bouts of fever (due to infections to which the severe anemia predisposes the child) and diarrhea and other intestinal problems.
What is Mediterranean anemia?
The gene for beta thalassemia is not evenly distributed among peoples. It is, for example, relatively more frequent in people of Italian and Greek origin, both of which are peoples from the Mediterranean. Because of this, thalassemia major has been called Mediterranean anemia.
The name thalassemia was coined at the University of Rochester in upstate New York by the Nobel Prize-winning pathologist George Whipple and the professor of pediatrics William Bradford from the Greek thalassa for sea and -emia, meaning the blood. Thalassemia means "sea in the blood." But for the Greeks, the sea was the Mediterranean, so thalassemia also conveys the idea of the Mediterrranean in the blood.
The reason that the gene for beta thalassemia is relatively common, for example, among people of Italian and Greek origin is that parts of Italy and Greece were once full of malaria. The presence of thalassemia minor (like sickle cell trait in Africa) afforded protection against malaria, and therefore, this gene thrived.
What is the genetic pattern of inheritance of beta thalassemia?
The pattern of genetic transmission of beta thalassemia (and sickle cell disease) was deciphered by James V. Neel when he was at the University of Rochester (he later went to Michigan). Dr. Neel recognized that the parents of children with thalassemia major had thalassemia minor with one beta thalassemia gene. When these parents had children, they have a 25% chance of having a thalassemia major child (with both genes for beta thalassemia), a 50% chance of having children with thalassemia minor (with only one gene for beta thalassemia), and a 25% chance of having a child without thalassemia major or minor (with both genes for normal beta chains). This form of inheritance is medically referred to as an autosomal recessive pattern.
The diagnosis of thalassemia major and minor
Persons with thalassemias have smaller sized red blood cells than normals as well as low red blood cell counts (anemia). Thalassemia major and thalassemia minor can now be diagnosed (and distinguished from one another) not only by conventional clinical and blood testing, but also by molecular medical tests. These tests permit accurate diagnosis to be made at any time, even before birth (in fact, well before the beta chain machinery is fired up to make beta chains for hemoglobin).
The treatment of thalassemia major
Infants with thalassemia major are well at birth because of a special form of hemoglobin present in the fetus and newborn. Eventually, however, this hemoglobin is replaced by defective hemoglobin. Symptoms emerge late in the first year of life. The child develops pale skin, irritability, growth retardation, swelling of the abdomen due to enlargement of the liver and spleen (hepatosplenomegaly) with jaundice. This is associated with severe anemia with rupture of the red blood cells (hemolytic anemia). The child with thalassemia major becomes dependent on blood transfusions and, although they do help, they create further problems including iron overload. Folic acid supplementation is often given. At this time, there is only treatment for relieving the symptoms of the illness for thalassemia major. Gene therapy remains a potential treatment for the future.
The long-term hope is that thalassemia major will be cured by insertion of the normal beta-chain gene through gene therapy or by another modality of molecular medicine.
Beta Thalassemia At A Glance
•There are two forms of beta thalassemia: thalassemia minor and thalassemia major (also called Cooley's anemia).
•Beta thalassemia is more frequent in people of Italian and Greek origin.
Reference:
Harrison's Principles of Internal Medicine,
McGraw-Hill, edited by Eugene Braunwald,
et. al., 2001.
Additional information is also available through the following organizations:
The National Institutes of Health (NIH)
Cellular Hematology Scientific Research Group
Blood Diseases Program
National Heart, Lung, and Blood Institute
6701 Rockledge Drive, MSC-7950
Bethesda, MD 20892-7950. USA
Phone: 301-435-0050
Fax: 301-480-0868
Cooley's Anemia Foundation,
129-09 26th Avenue - #203
Flushing, NY 11354, USA
Phone: 800-522-7222
Fax: 718-321-3340
Thalassemia International Federation
Philippou Hadjigerogiou No.1- Flat 8
P.O. Box 8807
Nicosia, Cyprus
Phone: (357) 2-319129
Fax: (357) 2-314552
Last Editorial Review: 12/11/2005
©1996-2010 MedicineNet, Inc. All rights reserved.
MedicineNet does not provide medical advice, diagnosis or treatment.
























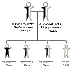












0 comments: